Czym jest CAH?
Wrodzony przerost nadnerczy, (CAH - congenital adrenal hyperplasia) związany jest z występowaniem bloków enzymatycznych na szlaku biosyntezy hormonów kory nadnerczy (glikokortykoidów oraz mineralokortykoidów). Zespól ten uwarunkowany jest genetycznie. Najczęściej niedobór enzymatyczny dotyczy 21-hydroksylazy, rzadziej 17-hydroksylazy, 11-hydroksylazy oraz dehydrogenazy 3ß-hydroksylowej. Niewystarczająca produkcja tych substancji powoduje zachwianie równowagi hormonalnej.
W rezultacie niskiego poziomu kortyzolu - nadnercza, pod wpływem gruczołów przysadkowych, zwiększają produkcję androgenów. W wyniku zmniejszenia produkcji kortyzolu kora nadnerczy, by wyrównać niedobór, produkuje za dużą ilość androgenów, czyli męskich hormonów sterydowych, które są niepożądane. Mówiąc w skrócie, podczas gdy jedna część nadnerczy wytwarza niedostateczną ilość kortyzolu i aldosteronu, druga część gruczołu produkuje zbyt dużo testosteronu. Ta ostatnia cecha odróżnia niedobór CAH-21-hydroksylazy od choroby Addisona, gdyż u osób cierpiących na to drugie schorzenie nadnercza zwykle w ogóle nie funkcjonują.
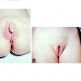
Klasyczny przerost nadnerczy ujawnia się po porodzie lub w dzieciństwie i obejmuje przede wszystkim objawy związane z zaburzeniami elektrolitowymi, występowaniem nadciśnienia tętniczego, a także maskulinizacje i hirsutyzm, obojnactwo płodów XX. Może być przyczyną różnicowania z zespołem policystycznych jajników, a wręcz tego wymaga - szczególnie tzw. opóźniony lub niepełny przerost nadnerczy (late-onset-CAH). Ujawnia się on w wieku późniejszym i nie towarzyszą mu zaburzenia w stężeniu mineralokortykoidów. Stwierdza się natomiast hirsutyzm, oligomenorrhoea, rzadziej przerost łechtaczki, łysienie.
4-6% pacjentek z objawami androgenizacji to późno ujawniający się wrodzony przerost nadnerczy. Wzrost stężenia testosteronu powyżej 2 ng/ml powinien skłonić do poszukiwania guza hormonalnie czynnego w obrębie jajników lub nadnerczy (oznaczenie 17-ketosterydów, 17-hydroksykosterydów, ocena USG jajników i nadnerczy).
Klasyczny niedobór CAH-21-hydroksylazy.
3/4 pacjentów z poważnym niedoborem zarówno kortyzolu jak i aldosteronu narażonych jest na nadczynność kory nadnerczy, powodującą odwodnienie i szok lub nawet śmierć, jeśli nie zostaną odpowiednio zdiagnozowane i leczone. U dzieci z klasycznym przypadkiem CAH-21, nadmierna produkcja androgenów w nadnerczach zaczyna się już we wczesnym okresie płodowym i powoduje nieprawidłowy przerost łechtaczki u dziewczynek i maskulinizację układu moczowo-płciowego.
Dziewczynki z pełno objawowym CAH często klasyfikowane są po porodzie jako chłopcy. Natomiast chłopcy z tym schorzeniem nie mają zniekształconych narządów płciowych w momencie urodzenia, a ciągły nadmiar androgenów powoduje atypowo szybki wzrost ciała. Zbyt wczesne pojawienie się symptomów dojrzewania płciowego, co powoduje, że dziecko zbyt wcześnie przestaje rosnąć i objawia się w przyszłości (u dorosłych) niskim wzrostem. Odpowiednie leczenie przywraca normalną równowagę hormonalną i umożliwia niemal normalny wzrost i prawidłowy cykl dojrzewania.
Odpowiednie zabiegi chirurgiczne wykonane przez doświadczonego pediatrę-urologa pozwalają odtworzyć kobiece narządy płciowe u noworodków żeńskich. Niektórzy z chirurgów są obecnie w stanie za jednym razem odtworzyć pochwę i zmniejszyć łechtaczkę we wczesnym okresie niemowlęctwa, podczas gdy dawniej był to proces, co najmniej dwustopniowy, który kończył się pod koniec okresu dojrzewania. Wciąż problematycznym pozostaje czas wykonywania procedur chirurgicznych na narządach płciowych noworodków żeńskich, gdyż zdarzają się przypadki dysforii płciowej u dorosłych osób poddanych takim zabiegom.
Nieklasyczny CAH - niedobór 21-hydroksylazy
Łagodniejsza, niepowodująca zagrożenia dla życia forma CAH-21 ujawnia się w późnej fazie dzieciństwa lub nawet w początkach dorosłości i nie charakteryzuje się odmienną budową narządów płciowych u dziewczynek. Osoby z tym typem CAH cechuje częściowy niedobór enzymów, a co za tym idzie lepsza niż w przypadku klasycznym produkcja kortyzolu, normalna produkcja aldosteronu i niższy poziom androgenów nadnerczowych. Osoby te nie są narażone na nadczynność nadnerczy. Ogólnie rzecz biorąc, pacjenci ci poszukują pomocy medycznej z powodu zbyt wczesnego rozwoju owłosienia łonowego, nieregularnych miesiączek, hirsutyzmu lub ciężkich przypadków trądziku. Niektóre z osób dotkniętych nieklasycznym CAH nie mają w ogóle symptomów i choroba zostaje u nich wykryta tylko dlatego, że występowała u któregoś z krewnych.
Rozpoznanie choroby - diagnoza
Rozpoznanie CAH opiera się tradycyjnie na pomiarze poziomu hormonów połączonym z obserwacją kliniczną, w skład, której wchodziła historia pacjenta (przypadki w rodzinie) i wieloaspektowa diagnostyka. W badaniach hormonalnych obserwuje się wzrost stężenia LH oraz mniejszy FSH, poziom testosteronu jest nieco podwyższony lub w normie, wzrasta natomiast poziom wolnego testosteronu, może wzrastać poziom DHEAS. Rozpoznanie stawia się na podstawie oznaczenia poziomu 17-hydroksyprogesteronu w surowicy. Wartości przekraczające 10 ng/ml pozwalają postawić rozpoznanie.
Gdy wartości mieszczą się w przedziale od 2 do 10 ng/ml należy w celu różnicowania wykonać test z ACTH. W przypadku wzrostu 3-krotnego poziomu 17-OH-progesteronu rozpoznajemy CAH. Leczenie polega na podawaniu dexometazonu, a przejściowo także leków antyandrogennych. Guzy wirilizujace nadnerczy i jajników występują rzadko i charakteryzują się nagłym początkiem i szybkim narastaniem hirsutyzmu oraz maskulinizacji.
W niektórych stanach USA oraz kilku innych krajach, w przeciągu kilku pierwszych dni po narodzeniu wykonuje się test hormonalny na obecność CAH. Bada się te same próbki krwi pobierane z pięty noworodka, które służą do rutynowej diagnostyki pourodzeniowej. Potrzebę wykonywania testów na obecność CAH u noworodków uzasadnia fakt, że śmiertelność spowodowana nadczynnością nadnerczy - głównie wśród noworodków płci męskiej, które nie mają zewnętrznych objawów choroby - jest wysoka, a zapobiec temu może wczesne zdiagnozowanie i szybko podjęte leczenie. Szacuje się, że na całym świecie 1 na 5,000 noworodków płci męskiej rodzi się z klasycznym CAH (lub 1 na 10 000 wszystkich noworodków), stąd wczesna diagnostyka przesiewowa ukierunkowana na CAH jest bardzo cenna - pozwala zapobiec zgonom dzieci płci męskiej obciążonych tym zespołem. Metody diagnostyczne ciągle ewoluują, zarówno, jeśli chodzi o metody hormonalne jak i nowsze badania genetyczne.
Dzięki rozpowszechnieniu technologii genetyki modularnej, możemy obecnie badać geny pacjentów z CAH oraz członków ich rodzin. Ten rodzaj badań ma zastosowanie w badaniach prenatalnych i noworodków, w poradnictwie genetycznym oraz przy potwierdzaniu diagnozy w niejasnych przypadkach. Diagnoza molekularna nie jest jeszcze dostępna jako test w zwykłych laboratoriach, można jednak te analizy wykonać w wyspecjalizowanych laboratoriach badawczo-genetycznych. Ponieważ badania DNA stają się coraz powszechniej akceptowane, bez wątpienia również te testy staną się bardziej dostępne. Badania genetyczne mogą pomóc w przypadku niejasności, związanych z testami hormonalnymi oraz umożliwiają dostarczenie rodzicom pacjentów z CAH dokładnych informacji na temat ryzyka urodzenia kolejnego dziecka z tą chorobą już w czasie pierwszych tygodni ciąży.
Winno się jednak z ostrożnością podchodzić do wykonywania testów genetycznych na masową skalę, po pierwsze z powodu możliwości złej interpretacji przez niedoświadczony personel, po drugie zaś z powodu niepokojącej możliwości użycia wyników tego testu przez pracodawców lub firmy ubezpieczeniowe. Należy zrozumieć, że nosiciele zmutowanego genu odpowiedzialnego za niedobór 21-hydroksylazy nie muszą jednocześnie mieć symptomów lub oznak CAH. Niektóre osoby mają 2 nieklasyczne, łagodne mutacje, a choroba dotyka ich tylko w minimalnym stopniu, nie powodując potrzeby medycznego bądź operacyjnego leczenia. Co więcej, istnieją również rzadkie przypadki osób z klasyczną mutacją genów, u których rozwinęła się osłabiona forma choroby.
Wszystkie omawiane testy muszą mieć ściśle poufny charakter. Testy genetyczne przypuszczalnie najlepiej sprawdzają się jedynie w przypadkach, kiedy dana osoba lub rodzina wykonuje je, by uzyskać wiedzę i podjąć ważne decyzję dotyczące leczenia oraz odpowiednio wczesnego przygotowania się na narodziny dziecka z CAH. Prawo dąży obecnie do zabezpieczenia przed nieuczciwym uzyskiwaniem i rozprzestrzenianiem informacji na temat chorób genetycznych.
Standardowe leczenie przypadków CAH:
W chwili obecnej standardowe leczenie medyczne polega na podawaniu glikokortykoidu, leku sterydowego podobnego do kortyzolu, na przykład doustnego hydrokortyzolu w przypadku dzieci oraz prednisonu lub dexometazonu w przypadku starszych pacjentów. Dodatkowo osoby z niedoborem aldosteronu w wyniku ciągłej utraty jonów sodowych (NaCl) potrzebują dodatkowego leku, fludrokortizonu (Florinef), który zastępuje aldosteron nie pozwalając na ucieczkę jonów sodowych.
Niemowlęta i małe dzieci mogą również przyjmować sól w tabletkach jako uzupełnienie diety, natomiast starsi pacjenci mogą spożywać posiłki bogate w sól. Pomimo tego, że wielu pacjentów dobrze znosi tego typu leczenie, trudno jest precyzyjnie naśladować prawdziwy rytm hormonalny i osiągnąć idealną równowagę hormonalną. Dlatego też większość pacjentów z CAH przechodzi przez przemienne okresy zachwiań równowagi hormonalnej istnieje, zatem konieczność częstego monitorowania laboratoryjnego (poziomy hormonów; w szczególności 17-hydroxyprogesteronu i androgenów), gdyż tak częste monitorowanie ich zawartości w organizmie pacjentów pozwala lekarzom na prawidłowe ustalenie oraz szybką modyfikację dawek leków.
Zaniedbanie w częstej diagnostyce prowadzi do zwiększenia dawek leków sterydowych, które czasem stają się zbyt duże. Znaną komplikacją spowodowaną dużymi dawkami glikokortykoidów jest zahamowanie wzrostu. Endokrynolodzy mogą poczuć się jak "między młotem a kowadłem" podczas leczenia niektórych pacjentów z CAH trudnym do kontroli, gdyż zarówno zbyt małe jak i zbyt duże dawki leków prowadzą w rezultacie do zahamowania wzrostu. Większość pacjentów z CAH nie osiąga wzrostu, jaki można było dla niego przewidzieć na podstawie wzrostu rodziców. Szczególnie ważnym czynnikiem determinującym ostateczny wzrost pacjentów z CAH jest ilość steroidów podawanych w pierwszych dwóch latach życia. Obecnie lekarze zalecają zmniejszone dawki leków, by zachować możliwość osiągnięcia większego wzrostu. Wszystkie dzieci z CAH powinny często odwiedzać pediatrę-endokrynologa, który nie tylko skontroluje poziom hormonów we krwi, ale również dokładnie zmierzy wzrost, wagę, ciśnienie krwi oraz raz w roku dokona prześwietlenia nadgarstka (zdjęcie rentgenowskie wieku kości).
Pacjenci z nieklasycznym CAH, jeśli wymagają terapii medycznej, zwykle skutecznie leczeni są przy użyciu jedynie hydrokortyzolu (dzieci) lub prednisonu (dorośli). Nie wymagają oni operacji narządów płciowych.
Nowsze metody postępowania:
Z powodu powyższych trudności z osiągnięciem dokładności w leczeniu ciężkiego CAH przy użyciu standardowej terapii, lekarze z Krajowego Instytutu Zdrowia w Bethesda w stanie Maryland, opracowali innowacyjny, eksperymentalny rodzaj terapii medycznej. Składa się na nią kombinacja 3 leków, z czynnikiem blokującym androgeny (Flutamid), hamującym produkcje aromatazy - enzymu odpowiedzialnego za wytwarzanie estrogenu z androgenów (Testolacton), oraz małej dawki hydrokortyzolu.
Ta nowa kombinacja porównywana jest ze standardową terapią, opisaną wcześniej, w losowo wybranych klinicznych przypadkach. Wstępne rezultaty po 6 miesiącach doświadczeń na małej grupie pacjentów są zachęcające - wzrost jest bardziej odpowiedni do wieku, a kości zrastają się odpowiednio wolniej w grupie eksperymentalnej. Eksperymentalna terapia jest w trakcie trwania, zatem o odległych skutkach przyjętego modelu farmakoterapeutycznego nie można jeszcze powiedzieć. Potrzeba wielu lat by w pełni ocenić bezpieczeństwo i skuteczność tej terapii, gdyż wielu pacjentów musi osiągnąć swój ostateczny wzrost by stwierdzić, czy utrzymane zostały długo- czy krótkoterminowe korzyści wynikające z tego leczenia.
W czasie, gdy dzieci będą przechodzić przez okres dojrzewania, naukowcy będą musieli określić, jak dostosować dawki flutamidu i testolactonu tak, by mógł nastąpić normalny wzrost oraz okres dojrzewania. Z lekami eksperymentalnymi wiążą się potencjalne efekty uboczne, pomimo iż w czasie początkowych 6 miesięcy żadne z nich nie zostały zauważone. Pod uwagę należy również wziąć fakt większych niedogodności dla pacjenta. Trudniej im jest pamiętać o braniu trzech leków trzy razy dziennie niż o braniu jednego bądź dwóch - dwa razy dziennie.
Bardziej radykalną propozycją alternatywnego leczenia CAH jest metoda chirurgiczna adrenalektomii. Terapia ta była w powszechnym użyciu w czasach zanim lekarze mieli dostęp do leków steroidowych. Obecnie zalecana jest ta metoda jedynie wybranym pacjentom, w szczególności tym, u których aktywność enzymów jest bardzo niska lub w ogóle nie występuje, w celu uniknięcia dużych dawek glikokortykoidów oraz/lub długotrwałego przyjmowania dużych dawek anty-androgenów.
Głównym powodem przemawiającym za tym typem operacji jest fakt, że obecnie można ją wykonać przy użyciu laparoskopii. Laparoskopia polega na wykonaniu jednego (bądź więcej) 1-calowego nacięcia, przez które wprowadza się przewód - zawierający sondę z otworem na narzędzia chirurgiczne. Przykładowo operacja wyrostka robaczkowego przy pomocy laparoskopii cechuje się mniejszą zachorowalnością po zabiegu oraz zmniejszeniem potencjalnych komplikacji w trakcie operacji. W przypadku dziewczynek z ciężkim przypadkiem CAH, adrenalektomia może być wykonywana jednocześnie ze zmniejszeniem łechtaczki oraz/lub odtworzeniem pochwy. Jak do tej pory nie było jednak nadzorowanych prób tego typu operacji. Oczywiste jest, że usunięcie obu nadnerczy prowadzi do choroby Addisona i w takim przypadku pacjent dalej musi przyjmować ekwiwalenty zarówno kortyzolu i aldosteronu.
Zwolennicy adrenalektomii wskazują na fakt, że zastępcze dawki hormonów w przypadku pacjentów z chorobą Addisona są mniejsze niż w przypadku pacjentów z CAH, a dzieci z chorobą Addisona nie cierpią na zatrzymanie wzrostu, nadwagę oraz maskulinizację i komplikacje w czasie okresu dojrzewania. Z drugiej strony nie wiemy jak wiele z ujemnych skutków nadmiernej produkcji androgenów w nadnerczach zostaje zaprogramowanych już w okresie płodowym i we wczesnym okresie niemowlęcym.
Terapia prenatalna
Terapia prenatalna stosowana jest od około dziesięciu lat. W rodzinach, w których wykryto CAH u jednego z dzieci, rodzice mogą korzystać z porad genetycznych, dzięki którym dowiedzą się, w jaki sposób choroba jest dziedziczona i jakie są możliwości wpływu na chorobę podczas następnych ciąży. Celem podawania dexometazonu kobietom w ciąży, które są w grupie ryzyka urodzenia kolejnego dziecka z CAH jest zmniejszenie wydzielania androgenów z gruczołów nadnerczy płodu płci żeńskiej, a co za tym idzie zmniejszenie ryzyka urodzenia dziewczynki z męskimi narządami płciowymi. Ponieważ wytwarzanie androgenów w nadnerczach rozpoczyna się w połowie lub pod koniec pierwszego trymestru zanim wykonuje się badania prenatalne, leczenie rozpoczyna się zanim jeszcze wiadomo, jakiej płci jest płód oraz czy ma CAH.
Ponieważ CAH jest chorobą recesywną, szanse na to, że dziecko odziedziczy zmutowany gen od każdego z rodziców-nosicieli wynosi 50%, a ryzyko urodzenia dziecka z CAH wynosi 25% w każdej ciąży. Natomiast z tego powodu, że tylko połowa z dzieci to dziewczynki, jedynie 1/8 płodów może skorzystać z leczenia prenatalnego. Dlatego też 7/8 płodów narażona jest na niepotrzebne przyjmowanie steroidów przez pępowinę. Kilkaset dzieci leczone było w ten sposób w okresie prenatalnym, a pobieżne obserwacje nie wykazały większych efektów ubocznych. Trudno oszacować, czy leczenie prenatalne zwiększa ryzyko poronienia, małej wagi w momencie narodzin lub łagodnych form uszkodzeń mózgu. Niedawny komentarz w JAMA (wydanie z 2 kwietnia 1997) zaleca ostrożne stosowanie prenatalnego leczenia deksometazonem i ścisłe nadzorowanie podawania leku przez komisje nadzorcze w szpitalu oraz komitety etyczne.
Niektóre z negatywnych opinii na temat terapii prenatalnej biorą się najprawdopodobniej z nierzetelnych międzygatunkowych badań robionych na gryzoniach oraz naczelnych, które odnosi się do ludzi. Mimo to należy z ostrożnością rozważyć możliwość długoterminowych, nierozpoznanych jeszcze komplikacji, które mogą być związane z użyciem terapii eksperymentalnych. Najbardziej przekonującym argumentem na ograniczenie terapii prenatalnej do badań naukowych jest fakt, że posiadamy niewielką ilość danych porównawczych dotyczących użycia glikokortykoidów w trakcie pierwszego trymestru ciąży i w gruncie rzeczy nie wszystkich długoterminowych efektów ubocznych.
Obecne i przyszłe kierunki badań:
W chwili obecnej większość pacjentów z CAH może być z pozytywnymi skutkami leczona w standardowy medyczny i chirurgiczny sposób. Diagnoza molekularna nie wpływa w znaczący sposób na sytuację pacjenta i ma głównie zastosowanie w badaniach naukowych lub przy badaniach prenatalnych. Ważne rezultaty diagnostyczne wykazują niskie koszta skryningu noworodków, co zapowiada szersze ich zastosowanie w przyszłości. Testy molekularno-genetyczne powinny zostać usprawnione poprzez rygorystyczną kontrolę jakości, co powinno z kolei doprowadzić do przyspieszenia ich wykonywania i obniżenia kosztów ich wykonania. Niejasności dotyczące leczenia, które należy wyjaśnić, dadzą odpowiedź na pytanie, czy nowe eksperymentalne terapie lekami lub adrenalektomia wpłyną znacząco na poprawę zdrowia pacjentów, czy też zastąpienie hormonów przez terapię genetyczną jest możliwym przełomem w leczeniu. Jak również pozostaje wciąż otwartym pytanie czy pourodzeniowa chirurgia narządów płciowych jest wyrazem etycznej troski o pacjenta, a może nadużyciem mającym źródła w przekonaniu, co do nieomylności medycyny w kształtowaniu płci?
Źródło: www.hermafrodyz.org
Zdjęcie: www.images-google.pl
 Strona główna
Strona główna Przeglądaj
Przeglądaj Profil
Profil Poczta
Poczta