Pierwotne chłoniaki skóry stanowią bardzo niejednorodną grupę nowotworów, charakteryzujących się klonalnym rozrostem limfocytów T, B, rzadziej komórek NK (natural killer), zapoczątkowanym w skórze i ograniczonym do jej zajęcia przez 6 mies. od rozpoznania. Definicja ta podkreśla odrębność między pierwotnymi chłoniakami skóry, a wtórnym zajęciem powłok skórnych w przebiegu chłoniaków węzłowych.
Wśród chłoniaków nieziarniczych pierwotne chłoniaki skóry zajmują drugie miejsce ze względu na częstość występowania, po pierwotnych chłoniakach przewodu pokarmowego. W Stanach Zjednoczonych odnotowuje się 0,5-1 nowych przypadków/100 tys. mieszkańców, ok. tysiąca nowych zachorowań rocznie. W Polsce brak jest dokładnych danych statystycznych na ten temat. Obserwacje prowadzone w klinikach dermatologicznych, hematologicznych i onkologicznych na terenie kraju potwierdzają jednak stopniowy wzrost zachorowań.
Unikalną cechą pierwotnych chłoniaków skóry, związaną z ich lokalizacją, jest dostępność zmian w badaniu klinicznym i możliwość łatwego pobierania wycinków ze zmian chorobowych do badań diagnostycznych, co umożliwia optymalne korelowanie stanu dermatologicznego i przebiegu klinicznego z obrazem histopatologicznym, immunofenotypem oraz zmianami molekularnymi. Szczególnego znaczenia w przypadkach pierwotnych chłoniaków skóry nabierają dane dotyczące okresów przeżycia. Odmienność w przebiegu klinicznym i rokowaniu tych chłoniaków, w porównaniu z ich węzłowymi odpowiednikami sprawia, że ta grupa nowotworów wymaga zupełnie odmiennego podejścia terapeutycznego.
Klasyfikacje chłoniaków nieziarniczych, obowiązujące do lat 90., oparte na kryteriach histologicznych, nie uwzględniały różnic w lokalizacji nowotworów, nie pozwalały tym samym na podkreślenie odrębności pierwotnych chłoniaków skóry i były częstą przyczyną złych rozpoznań i nieprawidłowego leczenia. W ostatnich trzech 10-leciach osiągnięcia immunologii i biologii molekularnej znalazły szerokie zastosowanie w badaniach nad chłoniakami skóry, co poszerzyło wiedzę na ich temat i przyczyniło się do wprowadzeniem nowych podziałów, uwzględniających ich specyfikę.
Od czasu opublikowania przez Jondala doniesienia poświęconego markerom powierzchniowym ludzkich limfocytów T, zastosowana przez niego metodyka z powodzeniem wprowadzona została do analizy nacieków limfocytarnych w rozrostach limforetikularnych skóry. Umożliwiło to wyodrębnienie dwóch zasadniczych grup pierwotnych chłoniaków skóry: wywodzących się z limfocytów B (cutaneous B-cell lymphoma, CBCL) oraz z limfocytów T (cutaneous T-cell lymphoma, CTCL). Okazało się, że większość chłoniaków zajmujących od początku rozwoju skórę, wywodzi się z linii limfocytów T, podczas gdy chłoniaki i białaczki nie naciekające skóry, to rozrosty złośliwe komórek B. Dalsze badania pozwoliły na stwierdzenie, iż mózgokształtne komórki charakterystyczne dla zespołu Sezary'ego oraz limfocyty występujące w naciekach skórnych w ziarniniaku grzybiastym to limfocyty T. W obu chorobach komórki nowotworowe prezentowały markery typowe dla komórek pochodzących z grasicy i lokalizowały się w skórze, co zasugerowało połączenie i zaszeregowanie obu tych jednostek do odrębnej grupy chłoniaków. Termin pierwotnych chłoniaków skóry T-komórkowych wprowadzony został po raz pierwszy przez Lutznera i wsp. w 1975 r. Dzisiaj wiadomo, że limfocyty odpowiedzialne za rozwój CTCL to skórne limfocyty T, stanowiące w warunkach prawidłowych ok. 15 proc. puli limfocytów krążących. Z czasem grupę CTCL powiększono o inne jednostki chorobowe, wcześniej nieznane lub klasyfikowane do innych grup schorzeń.
Badania prowadzone nad dobrze znanymi od wielu lat pierwotnymi chłoniakami skóry oraz analiza nowych jednostek, doprowadziły w 1997 r. do powstania klasyfikacji pierwotnych chłoniaków skóry, zaproponowanej przez Europejską Organizację do Badań i Leczenia Raka (European Organization for Research and Treatment of Cancer, EORTC). Głównym celem tego podziału, przedstawionego w tab. 1., było stworzenie klasyfikacji przydatnej klinicyście w codziennej pracy, a więc dostarczającej informacji pomocnych w określeniu przebiegu, rokowania i podjęcia właściwych metod leczenia. Klasyfikacja EORTC opiera się na równoczesnej analizie i ocenie danych klinicznych, histopatologicznych, immunohistochemicznych, genetycznych i molekularnych. W klasyfikacji nie używa się pojęć mało złośliwy i bardzo złośliwy chłoniak skóry w znaczeniu histologicznym, lecz wprowadza się ocenę przebiegu chłoniaka i kwalifikuje się przedstawione jednostki do trzech grup: o łagodnym, pośrednim i agresywnym przebiegu, wyodrębniając grupę jednostek tymczasowych, o rokowaniu nie w pełni sprecyzowanym.
Najczęściej występującą jednostką z grupy pierwotnych chłoniaków skóry jest ziarniniak grzybiasty (mycosis fungoides, MF), epidermotropowy CTCL, charakteryzujący się proliferacją małych i średnich limfocytów T z mózgokształtnymi jądrami. Pierwszy opis choroby podał Alibert w 1806r., zaś 70 lat później Bazin wyróżnił 3 klasyczne okresy MF: rumieniowy, zwany wcześniej okresem przedguzowatym, naciekowy i guzowaty. Ten charakterystyczny przebieg kliniczny jest niezbędny dla rozpoznania MF, natomiast postać pierwotnie guzowata, opisana przez Vidala i Brocqa, uważana jest dziś za inny niż MF, CTCL.
Etiologia MF nadal pozostaje niewyjaśniona. Rozwój nowotworu jest procesem wieloetapowym. Analiza klonalności poprzez badanie rearanżacji genu g receptora limfocytów T wykazała stopniowo występujące przemiany w trakcie progresji MF: od dominacji poliklonalnych limfocytów, wykrywanej w przyłuszczycy plackowatej wieloogniskowej, poprzez oligoklonalność, do dominacji limfocytów monoklonalnych, stwierdzanych w stadium guzowatym. Te przemiany powodowane są długotrwałymi stymulacjami. Hipoteza o wpływie czynników środowiskowych i zawodowych, takich jak ekspozycja na związki chemiczne i rozpuszczalniki, na rozwój MF nie została potwierdzona w badaniach kontrolowanych. Podobieństwo MF do białaczki T-komórkowej dorosłych, ściśle związanej etiologicznie z zakażeniem retrowirusami HTLV-I, skłoniło do podjęcia badań nad rolą tych wirusów w etiopatogenezie CTCL. Badania przeprowadzone w ciągu ostatnich lat w populacjach europejskich nie udowodniły bezpośredniej roli HTLV-I w patogenezie MF , mimo iż w pracach pochodzących z USA donoszono o wykrywaniu cząsteczek HTLV-I oraz sekwencji DNA wirusa w komórkach krwi obwodowej oraz wycinkach skórnych, pochodzących od chorych na MF. Nie można wykluczyć roli HTLV-I jako kofaktora w procesie nowotworowym.
Nie udowodniono także związku między zakażeniem wirusem Epstein-Barr a MF. Pierwotne chłoniaki skóry występują często w skojarzeniu z określonymi antygenami zgodności tkankowej: HLA Aw31, Aw32, B8, Bw35 i DR5. Przewlekła stymulacja może indukować aberracje genetyczne - kolejny krok w patogenezie pierwotnych chłoniaków skóry. Komórki pierwotnych chłoniaków skóry wykazują bardzo duże zdolności rozwoju wielu aberracji strukturalnych [25, 26]. Niestabilność genetyczna może się wiązać ze zmienioną aktywnością telomerazy, która - na ogół słaba - wzrasta nawet we wczesnych stadiach MF i w przyłuszczycy plackowatej. Rola onkogenów: p53 i białka bcl-2 w rozwoju CTCL jest nieznaczna.
Progresja nowotworu zależy od odpowiedzi immunologicznej skierowanej przeciw komórkom nowotworowym oraz od funkcjonowania sieci cytokin. W okresie rumieniowym MF w zmianach skórnych nie stwierdza się produkowanej przez limfocyty Th2 IL-4, w stadium naciekowym IL-4 występuje u części chorych, a w stadium guzowatym u większości chorych. Przyjmuje się, że rozrastające się w skórze nowotworowe limfocyty T produkują cytokiny typowe dla limfocytów Th2: IL-4, IL-5, IL-10; ponieważ interferon g produkowany jest przez limfocyty Th1, wykrywane we wczesnych stadiach MF, nienowotworowe limfocyty T naciekające skórę, należą do subpopulacji Th1.
MF występuje częściej u mężczyzn niż u kobiet. Choroba dotyczy głównie populacji dorosłych, u dzieci opisano tylko nieliczne przypadki.
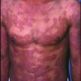
W początkowym stadium na skórze pojawiają się pojedyncze lub mnogie plamy rumieniowe różnej wielkości, niekiedy z delikatnym złuszczaniem na powierzchni. Często pierwsze wykwity zajmują skórę w miejscach osłanianych przed działaniem promieniowania słonecznego. Wykwitom może towarzyszyć świąd. Rokowanie w tej fazie jest bardzo indywidualne: opisywano całkowitą regresję zmian, jak i szybki postęp choroby. W okresie naciekowym do wykwitów plamistych dołączają się zmiany naciekowe i tarczki o różnych zarysach i wielkości, ostro odgraniczone od skóry zdrowej.
Średni czas przeżycia chorych z uogólnionymi zmianami rumieniowymi i naciekami wynosi ok. 12 lat. Pojawienie się na skórze guzów oraz owrzodzeń, będących zejściem wykwitów guzowatych, świadczy o progresji do okresu guzowatego. W każdym okresie rozwoju MF może wystąpić uogólniony rumień - erytrodermia, którą różnicuje się z zespołem Sezary'ego głównie na podstawie liczby atypowych limfocytów T w krwi obwodowej. Obok klasycznej formy MF znanych jest wiele odmian i wariantów tego chłoniaka: MF z towarzyszącą mucynozą mieszkową, MF w odmianie ziarniniakowej oraz ziarniniakowa obwisła skóra, ze zmianami pęcherzowymi czy odbarwionymi plamami. Ta różnorodność form klinicznych oraz fakt, iż chorobę poprzedzać mogą nieswoiste stany zapalne skóry sprawiają, że w wielu przypadkach MF prawidłowe rozpoznanie stawiane jest dopiero po kilku latach trwania choroby.
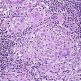
Obraz kliniczny w początkowym stadium MF przypomina dermatozy zapalne: przyłuszczycę plackowatą, wyprysk, łuszczycę czy atopowe zapalenie skóry. Zanim dojdzie do rozwoju typowego obrazu klinicznego, chorobę poprzedzać mogą nieswoiste stany zapalne skóry. Rozpoznanie MF opiera się głównie na badaniu histopatologicznym wycinków skórnych, które we wczesnym okresie rumieniowym sprawia istotne trudności interpretacyjne ze względu na nieswoisty obraz. Za najważniejsze kryteria w ocenie histopatologicznej wycinków skórnych, pozwalające na prawidłowe postawienie rozpoznania Smith uważa: pasmowaty naciek limfocytarny w górnej warstwie skóry i często dodatkowo występujące w brodawkach skórnych komórki nacieku zapalnego oraz epidermotropizm komórek jednojądrzastych.
W stadium rumieniowym epidermotropizm przejawiają najczęściej tylko pojedyncze komórki, zaś w dalszych okresach pojawiają się ich skupiska, w postaci tzw. mikroropni Pautrier. Część limfocytów naciekających skórę i naskórek wykazuje atypię komórkową i jądrową. Liczba komórek atypowych może być różna, niekiedy są one bardzo trudne do wykrycia. Zazwyczaj obserwuje są pojedyncze figury podziału mitotycznego. W jądrach występuje nadmiar chromatyny i pofałdowania błony jądrowej, co nadaje limfocytom mózgokształtny wygląd. Pozostałe cechy histopatologiczne są zmienne i odzwierciedlają obraz kliniczny zmian skórnych. Badanie wycinków skórnych w mikroskopie elektronowym ułatwia wykrycie pofałdowań błony jądrowej. Ocena morfometryczna pozwala na określenie tzw. indeksu konturu jądrowego (NCI), wyrażonego stosunkiem obwodu jądra do pierwiastka kwadratowego jego powierzchni. W przypadkach MF wartości NCI są znacznie wyższe niż w przypadkach łagodnych dermatoz, co wykorzystywane jest przez niektóre grupy badaczy w różnicowaniu tych jednostek. Metoda ta jest jednak stosunkowo mało czuła.
Badania cytofotometryczne DNA i analiza cytogenetyczna wzajemnie się uzupełniają, pozwalając na wykrycie nieprawidłowej liczby chromosomów w komórkach oraz precyzyjne określenie nieprawidłowości w poszczególnych chromosomach. Wprowadzone do diagnostyki liczne przeciwciała monoklonalne do wykrywania antygenów w materiale tkankowym zatopionym w parafinie, znacznie poszerzyły wartość badania immunofenotypów. Mnożące się w skórze limfocyty T mają najczęściej fenotyp CD4, a ściślej fenotyp limfocytów T pomocniczych indukujących (CD45RA-, CDW 29+) oraz główne markery dla limfocytów T (CD2, CD3, CD5); CD7 na ogół nie występuje. Dotyczy to okresu rumieniowego i naciekowego MF. W stadium guzowatym jeden lub więcej markerów komórkowych typowych dla limfocytów T zanika. Tylko w nieznacznej części przypadków MF występuje przewaga CD8 nad CD4. Immunofenotypowanie ma ograniczoną wartość w różnicowaniu wczesnych stadiów MF z łagodnymi dermatozami zapalnymi, a przyczyną jest prawdopodobnie zbyt mała liczba komórek nowotworowych, których nie wykrywa się na poziomie immunohistochemicznym. Metoda umożliwia monitorowanie procesu chorobowego i w pewnym stopniu także rokowanie; poprzez śledzenie markera CD 30 oraz określenie mnożącego się klonu limfocytów w skórze, węzłach chłonnych i krwi obwodowej. W zaawansowanym stadium MF nierzadko obserwuje się progresję do chłoniaka olbrzymiokomórkowego z limfocytów T, CD30+ lub CD30-, co wiąże się z agresywnym przebiegiem klinicznym.
Wprowadzenie do diagnostyki technik biologii molekularnej, umożliwiających badanie rearanżacji genów receptora limfocytów T, TCR (T-cell receptor), rozszerzyło możliwości wykrywania klonów rozrastających się limfocytów T. Wykrywanie klonów komórkowych prowadzono początkowo opierając się na technice Southern blot, jednak liczba komórek T w naciekach we wczesnych okresach MF może okazać się niewystarczająca do wykazania monoklonalności tą metodą. Częstość wykrywania monoklonalności znacznie zwiększyła się po wprowadzeniu do badań techniki PCR. Limfocyty T mogą prezentować 2 rodzaje łańcuchów ab i gd, co sprawia, że w badaniach diagnostycznych wykorzystywać można rearanżacje w obrębie czterech genów.
Większość skórnych limfocytów T wykazuje ekspresję ab TCR. Amplifikacja tych łańcuchów jest jednak dość trudna, z uwagi na dużą liczbę segmentów zmiennych, stąd w większości laboratoriów w celu wykrycia klonalności stosuje się amplifikację łańcucha g TCR. Złośliwa proliferacja zachodząca początkowo w skórze w miarę postępu choroby wykrywana jest również w węzłach chłonnych i krwi obwodowej. Wykrywanie rearanżacji genów TCR we krwi u pacjentów z agresywną postacią MF i zajęciem węzłów chłonnych stanowi potwierdzenie progresji choroby. Badania przy użyciu reakcji łańcuchowej polimerazy (PCR) prowadzone przez Wooda i wsp. pozwoliły na wykrycie dominującego klonu z rearanżacją genu łańcucha g TCR u 61 spośród 68 pacjentów z MF. Autorzy wykazali jednocześnie większą czułość techniki PCR w porównaniu z badaniami wykorzystującymi techniki Southern blot.
Bergman wykazał rearanżacje klonalne TCRg w naciekach skórnych u 69 proc. chorych z klasycznym MF, a Dadej w 88 proc. Muche i wsp. wykryli rearanżacje w limfocytach krwi obwodowej u 46,2 proc. chorych w stadium IA MF. Z punktu widzenia klinicznego i patologicznego pierwotne CTCL, zgodnie z definicją, rozwijają się w skórze i przez 6 mies. od początku choroby nie powinny dawać przerzutów do węzłów chłonnych czy narządów wewnętrznyc.
Badania z wykorzystaniem technik biologii molekularnej wykazały możliwość pozaskórnego rozsiewu nowotworowych limfocytów T już we wczesnym okresie choroby, o czym świadczy występowanie klonów komórek T we krwi obwodowej w stadiach IA i IB MF. Na częstość wykrywania klonów komórkowych w badanym materiale wpływają warunki techniczne, materiał wykorzystany do analizy oraz interpretacja wyników. Zwiększanie czułości stosowanych technik może jednak doprowadzić do wykrywania pojedynczych klonów reaktywnych limfocytów w zmianach skórnych w przebiegu niespecyficznych zmian zapalnych skóry. Badania Asthony-Keya i Bergmana udowodniły, że niespecyficzne zmiany zapalne skóry, w których występowały klonalne rearanżacje komórek T, mimo braku w momencie ustalania rozpoznania kryteriów klinicznych i histopatologicznych odpowiadających MF, po pewnym czasie uległy transformacji w MF. Dermatozy te określa się obecnie jako klonalne zapalenie skóry (clonal dermatitis) i pacjenci, u których rozpoznaje się tę jednostkę powinni być objęci obserwacjami klinicznymi i histopatologicznym, z uwagi na możliwość rozwoju CTCL.
Rozpoznanie MF powinno opierać się na wnikliwej ocenie stanu klinicznego chorych, badań histopatologicznych i immunohistochemicznych wycinków skórnych i węzłów chłonnych, analizie monoklonalności. Badania morfologiczne krwi obwodowej, szpiku, subpopulacji limfocytów krwi obwodowej, badanie radiologiczne klatki piersiowej oraz obrazowe narządów jamy brzusznej pozwalają określić rozległość procesu nowotworowego. Stadium zaawansowania klinicznego MF ocenia się na podstawie klasyfikacji opartej o system TNM, wprowadzonej przez Bunna i Lamberga, a zmodyfikowanej przez międzynarodową konferencję, określającą zasady leczenia chłoniaków skóry T-komórkowych.
Wybór metody leczenia zależy głównie od stadium MF, ale należy wziąć pod uwagę wiek i stan ogólny chorego, efekty uzyskane przy prowadzonym wcześniej leczeniu oraz możliwości terapeutyczne ośrodka podejmującego leczenie. W terapii MF wykorzystuje się wiele metod w różnych modyfikacjach. W przypadkach ograniczonych do powłok skórnych stosuje się silnie działające maści kortykostroidowe, chemioterapię miejscową chlormetyną albo karmustyną, fotochemoterapię (PUVA) bądź fototerapię UVB oraz radioterapię z zastosowaniem napromieniania całej skóry szybkimi wiązkami elektronów. Każdą z wymienionych metod można kojarzyć z podawaniem interferonów czy retinoidów. Ogólna polichemioterapia stosowana jest głównie jako leczenie paliatywne zaawansowanych przypadków MF, z zajęciem węzłów chłonnych czy narządów wewnętrznych lub w przypadkach nawrotów po wykorzystaniu innych metod leczenia. Agresywna chemioterapia stosowana w połączeniu lub jako jedyne leczenie nie poprawia okresów przeżycia, co skłoniło do stosowania metod mniej toksycznych. Leczenie ogólne prowadzi się początkowo w formie monoterapii kortykosteroidami, metotreksatem, lekami alkilującymi. Obok interefronów i retinoidów w leczeniu MF znalazły zastosowanie IL-2 i IL-12, cytokiny skoniugowane z toksynami, przeciwciała monoklonalne. Prowadzone są intensywne badania kliniczne nad szczepionkami, włączanymi do leczenia w przypadkach, w których inne metody zawiodły. Nadal jednak w stadiach zaawansowanych wyniki terapii są niezadowalające.
Źródło: www.termedia.pl
Zdjęcie: www.images.google.pl
 Strona główna
Strona główna Przeglądaj
Przeglądaj Profil
Profil Poczta
Poczta